Beta-Thalassämie ist eine angeborene Bluterkrankung, die durch Genmutationen verursacht wird, bei denen der Körper zu wenig Hämoglobin produziert - das Protein in roten Blutkörperchen, das Sauerstoff durch den Körper transportiert. In diesem Zustand werden auch rote Blutkörperchen zerstört. Im Laufe der Zeit kann unzureichendes Hämoglobin zu Anämie (niedrige Anzahl roter Blutkörperchen oder RBC) und anderen Komplikationen führen. Menschen mit der schwersten Form der Störung, Beta-Thalassämie major oder Cooley-Anämie, werden typischerweise mit regelmäßigen Bluttransfusionen behandelt. Bei Patienten mit dem kleineren Typ Beta Thalassemia intermedia sind möglicherweise weniger häufig Transfusionen erforderlich.
Violka08 / Getty ImagesTypen
Es gibt drei Arten von Beta-Thalassämie.
- Beta-Thalassämie-Merkmal, bei dem eine Person das Gen für die Störung trägt, aber keine Symptome aufweist. Es wird manchmal Thalassämie minor genannt.
- Beta-Thalassämie intermedia, eine relativ milde Form der Störung
- Beta-Thalassämie major oder Cooley-Anämie, die die schwerste Form darstellt
Beta-Thalassämie tritt am häufigsten bei Menschen mediterraner, asiatischer, indischer und afrikanischer Abstammung auf.
Symptome
Die Symptome einer Beta-Thalassämie hängen von der Art und dem Schweregrad der Störung ab. Menschen mit Beta-Thalassämie minor haben möglicherweise eine sehr leichte Anämie, entwickeln jedoch normalerweise keine erkennbaren Symptome. Einige wissen vielleicht nie, dass sie das veränderte Gen tragen.
Obwohl die beiden mutierten HBB-Gene bei der Geburt vorhanden sind, können Babys, die entweder mit Beta-Thalassämie intermedia oder Cooley-Anämie geboren wurden, erst zwischen 3 und 6 Monaten Symptome aufweisen. Einige Kinder mit Beta-Thalassämie entwickeln erst im Alter von 2 Jahren Symptome.
Unabhängig davon, wann ein Kind mit Beta-Thalassämie symptomatisch wird, tritt wahrscheinlich Folgendes auf:
- Anämie
- Ermüden
- Die Schwäche
- Kurzatmigkeit
- Gelbliche oder blasse Haut
- Langsames Wachstum
- Dunkler Urin
- Schlechter Appetit
- Umständlichkeit
Bei Patienten mit Cooley-Anämie können Symptome auftreten und Komplikationen auftreten, wie z.
- Abdominale Schwellung
- Knochendeformitäten oder Knochenbrüche aufgrund von Veränderungen im Knochenmark, in denen rote Blutkörperchen gebildet werden
- Splenomegalie, ein Zustand, bei dem die Milz, die rote Blutkörperchen filtert, überarbeitet ist und sich vergrößert
- Infektionen
Diagnose
Obwohl die meiste Zeit Thalassämie major auf dem Neugeborenen-Bildschirm identifiziert wird, können Menschen mit Thalassämie intermedia möglicherweise erst Jahre später identifiziert werden. Diese Personen werden im Allgemeinen anhand des routinemäßigen vollständigen Blutbilds (CBC) identifiziert. Die CBC zeigt eine leichte bis mittelschwere Anämie mit sehr kleinen roten Blutkörperchen. Dies kann mit Eisenmangelanämie verwechselt werden.
Die Diagnose wird durch ein Hämoglobinprofil (auch Elektrophorese genannt) bestätigt. Bei Beta-Thalassämie intermedia und Merkmal zeigt dieser Test eine Erhöhung des Hämoglobins A2 (eine zweite Form des adulten Hämoglobins) und manchmal des F (fötal). Alpha-Thalassämie intermedia wird im Allgemeinen als Hämoglobin-H-Krankheit bezeichnet, da dies das vorherrschende Hämoglobin im Profil ist.
Beta-Thalassämie kann durch verschiedene Blutuntersuchungen diagnostiziert werden. Eine mittelschwere und schwere Beta-Thalassämie wird normalerweise in der frühen Kindheit oder im Alter von 2 Jahren diagnostiziert, da Anzeichen und Symptome wie Anämie normalerweise sehr früh auftreten. Wenn Ihre Eltern an der Störung leiden, wurden Sie möglicherweise bei der Geburt oder als Kleinkind getestet und diagnostiziert, insbesondere wenn Sie in sehr jungen Jahren eine Anämie zeigten.
Die verschiedenen Tests zur Diagnose der Beta-Thalassämie umfassen:
- Das Neugeborenen-Screening, bei dem die meisten Säuglinge bei der Geburt routinemäßig getestet werden, um vererbte Zustände zu identifizieren. Dies kann eine Beta-Thalassämie oder zumindest Merkmale der roten Fahne der Störung im Blut zur weiteren Überwachung durch einen Hämatologen nachweisen. Beim Neugeborenen-Screening wird eine kleine Blutprobe entnommen und zur Auswertung an ein Labor geschickt.
- Ein vollständiges Blutbild (CBC), das das Hämoglobin sowie die Menge und Größe der roten Blutkörperchen misst. Die CBC zeigt auch eine leichte bis mittelschwere Anämie mit sehr kleinen roten Blutkörperchen, die typisch für Beta-Thalassämie sind. (Manchmal kann eine Diagnose jedoch mit einer Eisenmangelanämie verwechselt werden.)
Menschen mit milderen Formen der Thalassämie können diagnostiziert werden, nachdem eine routinemäßige Blutuntersuchung gezeigt hat, dass sie an Anämie leiden. Ärzte können auch Beta-Thalassämie bei Menschen afrikanischer, mediterraner oder ostasiatischer Abstammung vermuten und testen, die anscheinend an Anämie leiden.
- Eine Retikulozytenzahl, die die Gesundheit des Knochenmarks bestimmt und häufig Teil einer Aufarbeitung beim Testen auf Anämie ist, kann darauf hinweisen, dass das Knochenmark keine ausreichende Anzahl roter Blutkörperchen produziert.
- Die Überprüfung des Eisenspiegels im Blut zeigt, ob die Ursache der Anämie Eisenmangel oder Thalassämie ist.
Da es sich bei der Beta-Thalassämie um eine genetische Störung handelt, die vom Elternteil auf das Kind übertragen wird, können mehrere andere Tests das Vorhandensein des Gens und den möglichen Schweregrad der Beta-Thalassämie vorhersagen:
- Die Hämoglobin-Elektrophorese, eine Blutuntersuchung für Eltern, mit der verschiedene genetische Anomalien in der Hämoglobinstruktur festgestellt werden sollen, kann den Hämoglobinspiegel anzeigen.
- Pränatale Tests können den Schweregrad der Beta-Thalassämie in Fällen aufzeigen, in denen Eltern das Gen tragen. Dies können sein: Chorionzottenprobenahme in der 11. Schwangerschaftswoche, wobei ein winziges Stück der Plazenta zur Beurteilung entfernt wird; und Amniozentese, die etwa in der 16. Schwangerschaftswoche durchgeführt wird und die Untersuchung einer Probe der Flüssigkeit umfasst, die den Fötus umgibt.
Behandlung
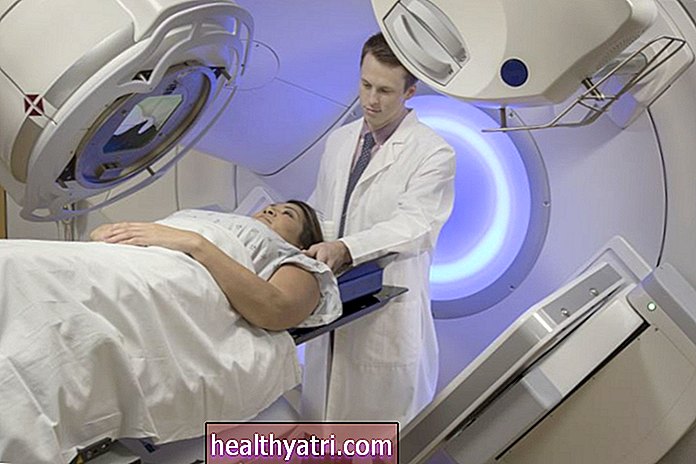
Die Standardbehandlungen für Menschen mit mittelschwerer oder schwerer Beta-Thalassämie sind Bluttransfusionen, Eisenchelat-Therapie und Folsäurepräparate:
- Bluttransfusionen: Eine Transfusion roter Blutkörperchen kann den Mangel ausgleichen und einen normalen Hämoglobinspiegel sicherstellen. Menschen mit Beta-Thalassämie major benötigen im Laufe ihres Lebens häufig alle zwei oder vier Wochen Transfusionen. Patienten mit Beta-Thalassämie intermedia benötigen möglicherweise nur gelegentliche Transfusionen während Krankheits- oder Pubertätsperioden.
- Eisenchelat-Therapie: Dies hilft dem Körper, überschüssige und möglicherweise schädliche Mengen an Eisen aus dem Blut zu entfernen - was aus vielen Transfusionen resultieren kann. Die Eisenchelatbildung wird oral oder durch eine Infusion unter die Haut verabreicht.
- Splenektomie: Die Anwendung dieses Verfahrens hat in den letzten Jahren abgenommen, aber die Entfernung der Milz wird manchmal in Betracht gezogen, wenn die Chelat-Therapie nicht funktioniert.
- Folsäurepräparate: Folsäure ist ein B-Vitamin, das beim Aufbau gesunder roter Blutkörperchen hilft. Ihr Arzt kann Folsäurepräparate zusätzlich zu Bluttransfusionen und Eisenchelat-Therapie empfehlen.
- Knochenmark- und Stammzelltransplantation: Eine Blut- und Markstammzelltransplantation ersetzt fehlerhafte Stammzellen durch gesunde Stammzellen einer anderen Person (eines Spenders). Stammzellen sind die Zellen im Knochenmark, die rote Blutkörperchen bilden. Obwohl dies eine mögliche Heilung für Beta-Thalassämie sein könnte, kann nur eine kleine Anzahl von Menschen mit schwerer Thalassämie eine gute Spenderübereinstimmung für dieses riskante Verfahren finden.
Menschen, die Beta-Thalassämie-Träger sind oder mit dem Merkmal identifiziert wurden, aber leichte oder gar keine Symptome haben, benötigen normalerweise nur sehr wenig Behandlung.
Ein Wort von Verywell
Wenn Sie oder Ihre Familienmitglieder an Beta-Thalassämie oder dem Gen für Beta-Thalassämie leiden, ist es eine gute Idee, mit Ihrem Arzt zu sprechen, wenn Sie über Kinder nachdenken. Ein genetischer Berater kann Ihnen dabei helfen, das Risiko zu bestimmen, dass die Störung an Ihre Nachkommen weitergegeben wird. Wenn Sie ein Baby erwarten und Sie und Ihr Partner Thalassämie-Träger sind, sollten Sie vorgeburtliche Tests in Betracht ziehen. Obwohl diese Tests zugegebenermaßen beängstigend und ahnungsvoll erscheinen, werden sie Ihre Angst lindern und Ihnen helfen, vorbereitet zu sein, unabhängig vom Ergebnis.
-count.jpg)